Imputation and Integration for Mouse Embryo data
loading package
[ ]:
import scanpy as sc
import torch as th
import scanpy as sc
import pandas as pd
import torch.nn as nn
from SpaMIE.create_graph import Sagegraph
from SpaMIE.spamie_main import Sagewrapper
device = th.device('cuda:0' if th.cuda.is_available() else 'cpu')
from matplotlib import rcParams
config = {
"font.family":'Times New Roman',
"font.size":12,
"axes.unicode_minus": False
}
rcParams.update(config)
Imputation
ATAC imputation RNA
[3]:
file_fold = '/data/xiangdw/data/data/MouseEmbryo/'
a = []
layers_nums = 3
for i in range(1):
seeds = i+1
adata_omics1 = sc.read_h5ad(file_fold + 'MouseEmbryo25um_ATAC.h5ad')
adata_omics2 = sc.read_h5ad(file_fold + 'MouseEmbryo25um_RNA.h5ad')
adata_omics1.obs['x_coord'] = adata_omics1.obs['row'].values
adata_omics1.obs['y_coord'] = adata_omics1.obs['col'].values
adata_omics1.obsm['spatial'] = adata_omics1.obs[['x_coord', 'y_coord']].values
adata_omics2.obs['x_coord'] = adata_omics2.obs['row'].values
adata_omics2.obs['y_coord'] = adata_omics2.obs['col'].values
adata_omics2.obsm['spatial'] = adata_omics2.obs[['x_coord', 'y_coord']].values
modalities = [adata_omics1, adata_omics2]
g_spatial_omics1, g_feature_omics1, adata_omics1, adata_omics2 = Sagegraph(modalities, device, datatype='Spatial-epigenome-transcriptome-ATAC-RNA', batch=False)
output_dir = '/data/xiangdw/data/pred result/'
weight = [0,0,1]
pred_name = 'mouseEmbryo_SpaMIE_RNA_'+str(layers_nums)+'_pred.csv'
true_name = 'mouseEmbryo_SpaMIE_RNA_'+str(layers_nums)+'_truth.csv'
in_feat = adata_omics1.obsm['feat'].shape[1]
out_feat = adata_omics2.X.shape[1]
model = Sagewrapper(seed=(int(seeds)), device=device, in_feat=in_feat, n_hidden=512, out_feat=out_feat, task='prediction', datatype='simu',
layers_nums=int(layers_nums), weight=weight, epoch=100, res_type='res_add', activation=nn.LeakyReLU
, sagetype='mean', lr=2e-3, lr2 = 0.001)
adata_omics1_pred, adata_omics2_pred, test_idx, train_idx,wt,alph = model.fit( adata_omics1, adata_omics2, g_spatial_omics1, g_feature_omics1,
output_dir=output_dir, pred_name=pred_name,
true_name=true_name, weight=True, save_csv=False)
/data/xiangdw/MODEL/SpaMIE/spamie_net.py:98: UserWarning: Implicit dimension choice for softmax has been deprecated. Change the call to include dim=X as an argument.
self.alpha = F.softmax(torch.squeeze(self.vu) + 1e-6)
RNA Impute ATAC
[4]:
file_fold = '/data/xiangdw/data/data/MouseEmbryo/'
a = []
layers_nums = 3
for i in range(1):
seeds = i+1
adata_omics1 = sc.read_h5ad(file_fold + 'MouseEmbryo25um_RNA.h5ad')
adata_omics2 = sc.read_h5ad(file_fold + 'MouseEmbryo25um_ATAC.h5ad')
adata_omics1.obs['x_coord'] = adata_omics1.obs['row'].values
adata_omics1.obs['y_coord'] = adata_omics1.obs['col'].values
adata_omics1.obsm['spatial'] = adata_omics1.obs[['x_coord', 'y_coord']].values
adata_omics2.obs['x_coord'] = adata_omics2.obs['row'].values
adata_omics2.obs['y_coord'] = adata_omics2.obs['col'].values
adata_omics2.obsm['spatial'] = adata_omics2.obs[['x_coord', 'y_coord']].values
modalities = [adata_omics1, adata_omics2]
g_spatial_omics1, g_feature_omics1, adata_omics1, adata_omics2 = Sagegraph(modalities, device, datatype='Spatial-epigenome-transcriptome-RNA-ATAC', batch=False)
output_dir = '/data/xiangdw/data/pred result/'
weight = [0,0,0]
pred_name = 'mouseEmbryo_SpaMIE_ATAC_'+str(layers_nums)+'_pred.csv'
true_name = 'mouseEmbryo_SpaMIE_ATAC_'+str(layers_nums)+'_truth.csv'
in_feat = adata_omics1.obsm['feat'].shape[1]
out_feat = adata_omics2.X.shape[1]
model = Sagewrapper(seed=(int(seeds)), device=device, in_feat=in_feat, n_hidden=512, out_feat=out_feat, task='prediction', datatype='simu',
layers_nums=int(layers_nums), weight=weight, epoch=300, res_type='res_add', activation=nn.LeakyReLU
, sagetype='gcn', lr=1e-4, lr2 = 0.001)
adata_omics1_pred, adata_omics2_pred, test_idx, train_idx,wt,alph = model.fit(adata_omics1, adata_omics2, g_spatial_omics1, g_feature_omics1,
output_dir=output_dir, pred_name=pred_name,
true_name=true_name, weight=True, save_csv=False)
/data/xiangdw/conda_env/GNNS/lib/python3.8/site-packages/scanpy/preprocessing/_highly_variable_genes.py:61: UserWarning: `flavor='seurat_v3'` expects raw count data, but non-integers were found.
warnings.warn(
/data/xiangdw/MODEL/SpaMIE/spamie_net.py:98: UserWarning: Implicit dimension choice for softmax has been deprecated. Change the call to include dim=X as an argument.
self.alpha = F.softmax(torch.squeeze(self.vu) + 1e-6)
Integration
[ ]:
import os
import dgl
import pandas as pd
import sys
import scanpy as sc
import importlib
import torch as th
import torch.nn as nn
from model_integration import *
from matplotlib import rcParams
config = {
"font.family":'Times New Roman',
"font.size":20,
"axes.unicode_minus": False
}
rcParams.update(config)
/data/xiangdw/MODEL
[ ]:
import torch.nn.functional as F
from SpaMIE.create_graph import Sagegraph
from SpaMIE.spamie_main import Sagewrapper
import numpy as np
from model_integration import set_seed
device = th.device('cuda:1' if th.cuda.is_available() else 'cpu')
for i in range(1):
seeds = str(i+1)
path = '/data/xiangdw/data/data/MouseEmbryo/'
adata_omics1 = sc.read_h5ad(path + 'MouseEmbryo25um_ATAC.h5ad')
adata_omics2 = sc.read_h5ad(path + 'MouseEmbryo25um_RNA.h5ad')
adata_omics1.obs['x_coord'] = adata_omics1.obs['row'].values
adata_omics1.obs['y_coord'] = adata_omics1.obs['col'].values
adata_omics1.obsm['spatial'] = adata_omics1.obs[['x_coord', 'y_coord']].values
adata_omics2.obs['x_coord'] = adata_omics2.obs['row'].values
adata_omics2.obs['y_coord'] = adata_omics2.obs['col'].values
adata_omics2.obsm['spatial'] = adata_omics2.obs[['x_coord', 'y_coord']].values
set_seed(2024)
modalities = [adata_omics1, adata_omics2]
g_spatial_omics1, g_feature_omics1, g_spatial_omics2, g_feature_omics2, adata_omics1, adata_omics2 = Sagegraph(modalities, device, task='Integration',
datatype="Spatial-epigenome-transcriptome-RNA-ATAC",batch=False)
in_feat = adata_omics1.obsm['feat'].shape[1]
out_feat = adata_omics2.X.shape[1]
weight = [1,1,1]
model = Sagewrapper(seed=(int(seeds)), device=device, in_feat=in_feat, n_hidden=256, out_feat=out_feat, task='integration', datatype='simu',
layers_nums=int(3), weight=weight, epoch=600, res_type='res_add', activation=nn.LeakyReLU
, sagetype='mean', lr=2e-4, lr2 = 0.002)
output = model.fit(adata_omics1, adata_omics2, g_spatial_omics1, g_feature_omics1,
g_spatial_omics2, g_feature_omics2, weight_factors=[3,1,1,1])
adata_omics2.obsm['SpaMIE'] = output[0].detach().cpu().numpy()
/data/xiangdw/conda_env/GNNS/lib/python3.8/site-packages/scanpy/preprocessing/_highly_variable_genes.py:61: UserWarning: `flavor='seurat_v3'` expects raw count data, but non-integers were found.
warnings.warn(
/data/xiangdw/MODEL/SpaMIE/spamie_net.py:98: UserWarning: Implicit dimension choice for softmax has been deprecated. Change the call to include dim=X as an argument.
self.alpha = F.softmax(torch.squeeze(self.vu) + 1e-6)
/data/xiangdw/MODEL/SpaMIE/spamie_net.py:66: UserWarning: Implicit dimension choice for softmax has been deprecated. Change the call to include dim=X as an argument.
self.alpha = F.softmax(torch.squeeze(self.vu) + 1e-6)
[ ]:
from SpatialGlue.utils import clustering
tool = 'louvain'
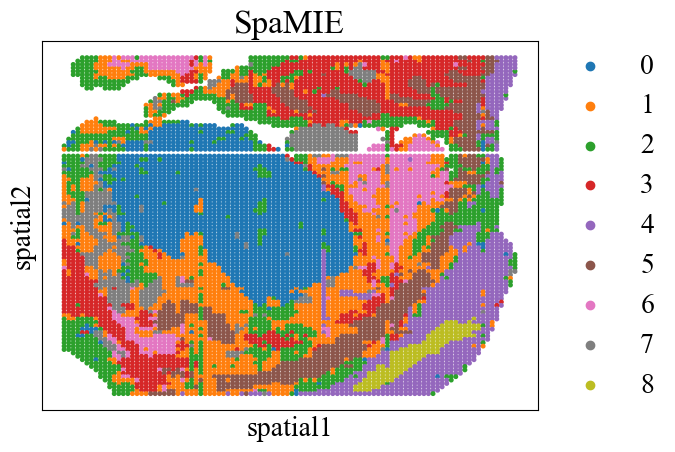
clustering(adata_omics2, key='SpaMIE', add_key='SpaMIE',start=0.001, end=1.0, increment=0.05, n_clusters=9, method=tool, use_pca=False)
sc.pl.embedding(adata_omics2, basis='spatial', color=['SpaMIE'], title='SpaMIE', s=50, show=False)
Searching resolution...
resolution=0.9510000000000001, cluster number=11
resolution=0.901, cluster number=9
/data/xiangdw/conda_env/GNNS/lib/python3.8/site-packages/scanpy/plotting/_tools/scatterplots.py:394: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
<Axes: title={'center': 'SpaMIE'}, xlabel='spatial1', ylabel='spatial2'>